
11: Transcription: Promoters, terminators and mRNA
- Page ID
- 380

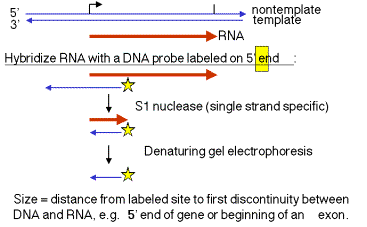
1. "S1 protection assay"
This assay measures the distance between an end label (at a specific known site on DNA) and the end of a duplex between RNA and the labeled DNA. A fragment of DNA (complementary to the RNA) that extends beyond the 5' end of the RNA is labeled at a restriction site within the RNA‑complementary region. The labeled DNA is hybridized to RNA and then digested with the single‑strand specific nuclease S1. The resulting fragment of protected DNA is run on a denaturing gel to determine its size. Note that this fragment runs from the labeled site to the nearest interruption between the DNA and the RNA. This could be the beginning of the RNA, or it could be an intron, or it could be an S1 sensitive site.
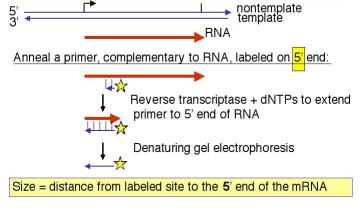
3. How do you label DNA at the ends?
- 5' end label: T4 polynucleotide kinase and [g 32P] ATP. The reaction is most efficient if the 5' phosphate is removed (by alkaline phosphatase) prior to the kinase treatment.
- 3' end label: Klenow DNA polymerase plus [a 32P] dNTP. The labeled dNTP is chosen to be complementary to the first position past the primer. A restriction fragment with a 5' overhang is ideal for this "fill‑in" labeling.
- Digestion with a second restriction endonuclease will frequently work to remove the label at the "other" end. One can also use electrophoretic gels that separate strands.
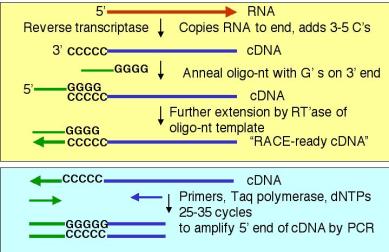
4. A PCR-based technique to determine the 5’ ends of mRNAs and genes
B. General methods for identifying the site for sequence‑specific binding proteins
1. Does a protein bind to a particular region?
a. Electrophoretic mobility shift assay (EMSA), or gel retardation assay: This assay will test for the ability of a particular sequence to form a complex with a protein. Many protein‑DNA complexes are sufficiently stable that they will remain together during electrophoresis through a (nondenaturing) polyacrylamide gel. A selected restriction fragment or synthetic duplex oligonucleotide is labeled (to make a probe) and mixed with a protein (or crude mixture of proteins). If the DNA fragment binds to the protein, the complex will migrate much slower in the gel than does the free probe; it moves with roughly the mobility of the bound protein. The presence of a slowly moving signal is indicative of a complex between the DNA probe and some protein(s). By incubating the probe and proteins in the presence of increasing amounts of competitor DNA fragments, one can test for specificity and even glean some information about the identity of the binding protein.
Figure 3.2.4. Diagram of results from an electrophoretic mobility shift assay

In this example, two proteins recognize sequences in the labeled probe, forming complexes A and B (lane 2). The proteins in complexes A and B recognize specificDNA sequences in the probe. This is shown by the competition assays in lanes 3-8. An excess of unlabeled oligonucleotide with the same sequence as the labeled probe (“self”) prevents formation of the complexes with labeled probe, whereas “nonspecific DNA” in the form of E. coli DNA does not compete effectively (compare lanes 6-7 with lanes 3-5).
This experiment also provides some information about the identity of the protein forming complex A. It recognizes an Sp1-binding site, as shown by the ability of an oligonucloetide with an Sp1-binding to compete for complex A, but not complex B (lanes 9-11). Hence the protein could be Sp1 or a relative of it.. The proteins forming complexes A and B do not recognize an Oct1-binding site (lanes 12-14).
b. Nitrocellulose binding: Free duplex DNA will not stick to a nitrocellulose membrane, but a protein‑DNA complex will bind.
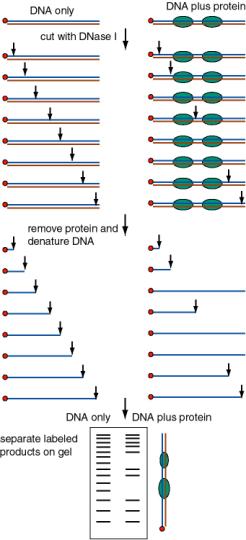
2. To what sequence in the probe DNA is the protein binding?
The presence of a protein will either protect a segment of DNA from attack by a nuclease or other degradative reagent, or in some cases will enhance cleavage (e.g. to an adjacent sequence that is distorted from normal B‑form). An end‑labeled DNA fragment in complex with protein is treated with a nuclease (or other cleaving reagent), and the protected fragments are resolved on a denaturing polyacrylamide gel, and their sizes measured.
- a. Exonuclease protection assay: The protein will block the progress of an exonuclease, so the protected fragment extends from the labeled site to the edge of the protein furtherest from the labeled site. One can use a combination of a 3' to 5' exonuclease (ExoIII) and a 5' to 3' exonuclease (l exonuclease) to map both edges.
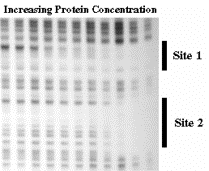
- b. DNase footprint analysis: DNase I will cut at many (but not all) phosphodiester bonds in the free DNA. The protein‑DNA complex is treated lightly with DNase I, so that on average each DNA molecule is cleaved once. The presence of a bound protein will block access of the DNase, and the bound region will be visible as a region of the gel that has no bands, i.e. that was not cleaved by the reagent.
4. DNA sequence‑affinity chromatography to purify DNA binding proteins
The specific binding sites (often 6 to 8 bp) can serve as an affinity ligand for chromatography. Multimers of the binding site are made by ligating together duplex oligonucleotides that contain the specific site. After a few crude initial steps (e.g. isolating all DNA‑binding proteins on DNA‑sepharose) the extract is applied to the affinity column. Most of the proteins do not bind, and subsequently the specifically bound proteins are eluted.
C. Promoters and the Initiation of Transcription: General Properties
- A promoter is the DNA sequence required for correct initiation of transcription
- Phenotype of promoter mutants
a. cis‑acting: A cis-acting regulatory element functions as a segment of DNA to affect the expression of genes on the same chromosome that it is located on. Cis-acting elements do not encode a diffusible product. The promoter is a cis-acting regulatory element.
Compare the phenotypes of mutations in the gene encoding b‑galactosidase (lacZ) versus mutations in its promoter (p).
Consider a heterozygote that is p+ lacZ‑ /p+ lacZ+ .
The phenotype is Lac+. lacZ+ complements lacZ‑ in trans. In this case, lacZ+ is dominant to lacZ-.
Consider a heterozygote that is p+ lacZ‑ /p‑ lacZ+ .
The phenotype is Lac‑. p+ does not complement p‑ in trans.
p‑ operates in cis to prevent expression of lacZ+ on this chromosome. The mutant promoter is dominant over the wild-type when the mutant promoter is in cisto the wt lacZ.
Consider a heterozygote that isp+ lacZ+ /p‑ lacZ‑ .
The phenotype is Lac+. lacZ+ now complements lacZ‑ in trans because it is driven by a functional promoter in cis, p+
b. Dominance in cis: the promoter “allele” that is in cisto the wild-type structural gene (lacZ) is dominant over the other promoter allele.
c. Promoter mutations affect the amount of product from the gene but do not affect the structure of the gene product.
D. Bacterial promoters
- Bacterial promoters occur just 5' to and overlap the start site for transcription(usually)
- Bacterial promoters are the binding site for E. coliRNA polymerase holoenzyme. The promoter covers about 70 bp from about ‑50 to about +20.
- Consensus sequences in the E. colipromoter
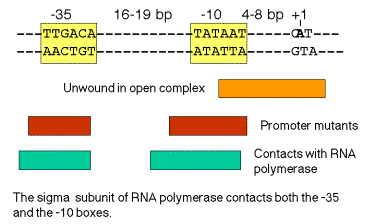
a. ‑35 and ‑10 sequences
‑35 16‑19 bp ‑10+1
‑‑‑‑‑‑‑‑TTGACA‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑--TATAAT‑‑‑CAT
Recognition by Allows binary complex to convert
RNA polymerase from closed to open
holoenzyme
b. The sequences are conserved in all E. coli genes transcribed by holoenzyme with s70
4. Promoter mutants
- a. Tend to fall into or close to one of these hexanucleotides
- b. Affect the level of gene expression, not the structure of the gene product
- c. Down promoter mutations: decrease the level of transcription. Tend to make the promoter sequence less like the consensus.
- d. Up promoter mutations: increase the level of transcription. Tend to make the promoter sequence more like the consensus.
- e. Down promoter mutations in the ‑35 sequence: decrease the rate of formation of the closed complex, indicating this is the sequence needed for intial recognition by the polymerase holoenzyme.
- f. Down promoter mutations in the ‑10 sequence: decrease the rate of conversion from the closed to the open complex, again supporting the proposed role for this A+T rich hexanucleotide.
- g. The critical contact points between RNA polymerase and the promoter tend to be in or immediately upstream from the consensus ‑35 and ‑10 boxes. (See Figure 3.2.7). Thus the biochemical and genetic data all support the importance of these conserved sequences.
Figure 3.2.7. Correlation of conserved sequences, location of promoter mutants, and regions of contact with polymerase at bacterial promoters
5. Alternate s factors can control the expression of sets of genes
- a. Alternative s factors make complexes with the core polymerase to direct the new holoenzyme to a particular set of promoters that differ in sequence from the general E. coli promoter sequence. Thus the polymerase can be directed to trancribe a new set of genes. This is one way to control gene expression.
- b. Examples include s factors for heat‑shock response (s32), transcription of genes involved in chemotaxis and flagellar formation (s28), and nitrogen starvation (s54). The s factors are named by their size in kDa.
- c. Three of the E. colis factors have regions of sequence similarity (s70,s32, and s28 ) whereas s54 is a distinctly different molecule that works rather differently.
| Factor | Gene | Use | ‑35 | Separation | ‑10 |
| s70 | rpoD | General | TTGACA | 16-19 bp | TATAAT |
| s32 | rpoH | Heat shock | CCCTTGAA | 13-15 bp | CCCGATNT |
| s28 | fliA | Flagella | CTAAA | 15 bp | GCCGATAA |
| s54 | rpoN | Nitrogen starvation | CTGGNA | 6 bp | TTGCA |
E. Promoters for eukaryotic RNA polymerases
Promoters contain binding sites for nuclear proteins, but which of these binding sites have a function in gene expression? This requires a genetic approach for an answer.
1. Use of "surrogate genetics" to define the promoter
a. In vitro mutagenesis (deletions or point mutations)
- Mutations of the binding sites for activator proteins lead to a decrease in the level of transcription of the gene. [Loss of function].
- Addition of a DNA fragment containing these binding sites will activate (some) heterologous promoters. [Gain of function].
- Sequences of the binding sites are frequently well conserved in promoters for homologous genes from related species.
- A potential regulatory region is initially examined by constructing progressive deletions from the 5' end (with respect to the direction of transcription) and also from the 3' end. Subsequently one can make clusters of point mutations (e.g. by linker scanning mutagenesis) or individual point mutations.
b. Test in an expression assay
(1)The mutagenized promoter is linked to a reporter gene so that RNA or protein from that gene can be measured quantitatively
- (a) Gene itself ‑ measure RNA production by S1 protection, primer extension, or other assay that is specific for a particular RNA
- (b) Heterologous reporter gene: encodes an enzyme whose activity is easy to measure quantitatively. Note that these measures of expression require both transcription and translation, in contrast to measurement of RNA directly. E.g., the genes encoding:
- b‑galactosidase: colorimetric assay, monitor the cleavage of o‑nitrophenyl‑b‑galactoside
- chloramphenicol (Cm) acetyl transferase (CAT): measure the acetylation of Cm, ususally use [14C] Cm; this is the enzyme that confers resistance to Cm in bacteria
- luciferase: monitor the emmission of photons resulting from the ATP‑dependent oxidation of luciferin; this is the enzyme that catalyzes light production in firefly tails
(2) The promoter‑reporter DNA constructs are introduced into an assay system that will allow the reporter to be expressed.
(a) Whole cells
microinjection into Xenopus oocytes
transfection of cell lines: introduce the DNA via electroporation or by getting the cells to take up a precipitate of DNA and Ca phosphate by pinocytosis
(b) Whole animals = transgenic animals
Introduce the DNA into the germ line of an animal, in mammals by microinjecting into a fertilized egg and placing that into a pseudopregnant female. This technology allows one to examine the effects of the mutation throughout the development of the animal.
(c) Cell‑free systems
Extracts of nuclei, or purified systems (i.e. with all the necessary components purified)
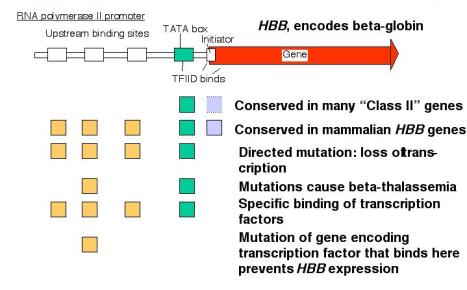
2. Promoter for RNA Pol II
a. The minimal promoter is needed for basal activity and accurate initiation.
- Needed for assembly of the initiation complex at the correct site
- DNA sequences
(a) TATA box
- Initially identified as a well conserved sequence motif about 25 bp 5' to the cap site (The cap site is the usual start site for transcription)
- The transcription factor TFIID binds to the TATA box
- Mutations at the TATA box generates heterogeneous 5' ends of the mRNAs ‑ indicative of a loss of start site specificity
(b) Initiator
- Sequences at the start site for transcription have consensus YANWYY (Y = C or t, W = T or A)
- Mode of action is still under investigation. Recent data indicate that TFIID also binds to the initiator; binds to one of the TAFs (see below).
- TATA plus initiator is the simplest minimal promoter.
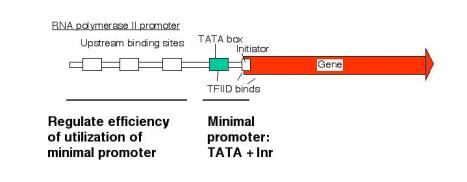
b. The amount of expression is regulated via upstream elements.
- Proteins bind to specific sequences (usually) 5' to the TATA box to regulate the efficiency of utilization of the promoter.
- These are frequently activators, but proteins that exert negative control are also being characterized.
- Examples of activator proteins
Sp1: binds GGGGCGGGG = GC box
Octn: binds ATTTGCAT = octamer motif
Oct1 is a general factor (ubiquitous)
Oct2 is specific for lymphoid cells
CP1, CTF = NF1, C/EBP bind to CCAAT = CCAAT box (pronounced "cat" box)
These are different families of proteins, CP1 and CTF are found in many cell types, C/EBP is found in liver and adipose tissue.
(4) These upstream control elements may be inducible (e.g. by hormones), may be cell‑type specific, or they may be present and active in virtually all cell types (i.e. ubiquitous and constitutive).
Figure 3.2.10.

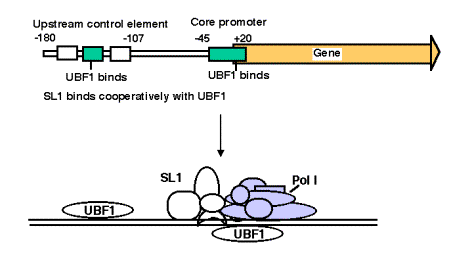
3. Promoter for RNA Pol I
- a. The core promoter covers the start site of transcription, from about ‑40 to about +30. The promoter also contains an upstream control element located about 70 bp further 5', extending from ‑170 to ‑110.
- b. The factor UBF1 binds to a G+C rich sequence in both the upstream control element and in the core promoter. A multisubunit complex called SL1 binds to the UBF1‑DNA complex, again at both the upstream and core elements. One of the subuntis of SL1 is TBP.
- c. RNA polymerase I then binds to this complex of DNA+UBF1+SL1 to initiate transcription at the correct nucleotide and the elongate to make pre‑rRNA.
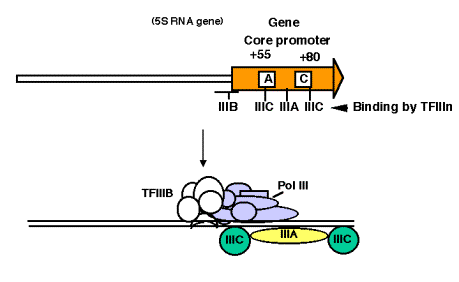
4. Promoter for RNA Pol III
a. This promoter has internal control sequences. Deletion of 5' flanking DNA still permits efficient transcription of (most) genes transcribed by RNA PolIII. Even the intial part of the gene is expendable, as is the 3' end. Sequences internal to the gene (e.g. +55 to +80 in 5S rRNA genes) are required for efficient initiation, in contrast to the familiar situation in bacteria, where most of the promoter sequences are 5' to the gene.
F. Enhancers
- Enhancers are DNA sequences that cause an increase in the level of expression of a gene with an intact promoter. They may act to increasethe efficiency of utilization of a promoter, or they may increase the probability that a promoter is in a transcriptionally competent chromatin conformation. This will be explored further in Part Four.
- They are operationally defined by their ability to act in either orientation and at a variety of positions and distances from a gene, i.e. act independently of orientation and position. This contrasts with promoters, that act (usually) in only one orientation and (usually) are at or close to the 5' end of the gene.
- They consist of binding sites for specific activator proteins. Always have multiple binding sites, often for several different activator proteins.
- Particular sets of genes can be regulated by their need for defined sets of activator proteins at their enhancers.
G. Elongation of transcription
- RNA polymerase must be released from the initiation complex to transcribe the rest of the gene. Elongation must be highly processive, i.e. once the polymerase begins elongation, it must transcribe that template all the way to the end of the gene.
- The factors required for initiation are not needed (and may inhibit) elongation, and they dissociate.

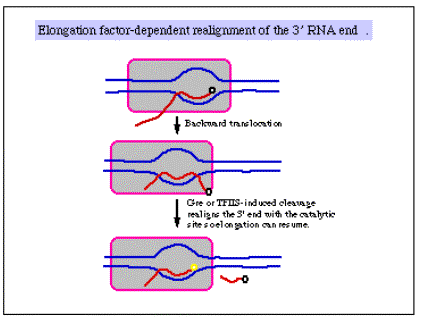
3. There is some indication that factors that increase the processivity of the transcription complexbind to the elongating polymerase. Examples include the following.
- NusA in bacteria
- GreA and GreB in bacteria
- TFIIS in eukaryotes, possibly many others.

a. r‑independent sites [Note: r = rho]
- Identified in vitro
- G+C rich hairpin followed by about 6 U's
- Hairpin is thought to be a site at which RNA polymerase pauses, and the weak rU‑dA base pairs in the RNA‑DNA heteroduplex allow melting of the duplex and termination.
- Some of the best examples of r-independent terminators are integral parts of the mechanism of regulation. Examples include the attenuators in the trpoperon and other amino acid biosynthetic operons. The r-independent terminators may be a specialized adaptation for regulation.
b. r‑dependent sites
- C‑rich, G‑poor stretch
- Requires the action of the protein r both in vitro and in vivo
- The r-dependent terminators are used at the 3' ends of many eubacterial genes.
2. r factor
- a. Hexamer, each subunit 46 kDa
- b. RNA‑dependent ATPase
- c. Gene for r is essential for E. coli
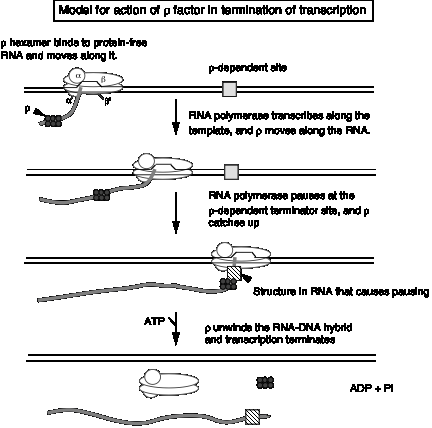
3. Model for action of r factor
- a. r binds to protein‑free RNA and moves along it
- b. When it reaches a paused polymerase, it causes the polymerase to dissociate and unwinds the RNA‑DNA duplex, thereby terminating transcription. This last step utilizes the energy of ATP hydrolysis. The protein r serves as the ATPase.
Figure 3.2.18.
I. Termination of transcription in eukaryotes
1. Termination by RNA Pol II
- a. No clear evidence for a discrete terminator for RNA polymerase II
- b. 3' end of mRNA is generated by cleavage and polyadenylation
- c. Signal for cleavage and polyadenylation:
(1) AAUAAA, about 20 nt before the 3' end of the mRNA
(2) Other sequences 3' to cleavage site
- d. Cleavage enzyme not well characterized at this point; the U4 snRNP may play a role in cleavage. A polyA polymerase has been identified.
- e. Polyadenylation is required for termination by RNA Pol II; possibly also pausing by the RNA polymerase
J. mRNA structure in bacteria
1. Bacterial mRNA is often polycistronic.

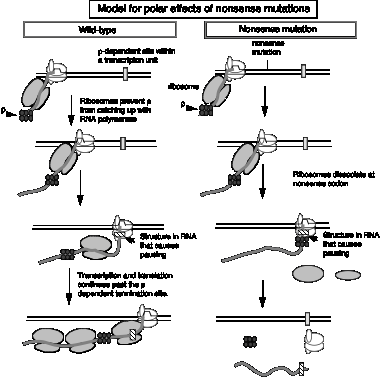
2. Model for r action can explain why stopping translation can also lead to a cessation of transcription.
- a. Suppose a r‑dependent terminator of transcription is present in the first gene of an operon. Normally it does not cause transcription to stop because it is covered by ribosomes translating the mRNA, and the subsequent genes in the operon are transcribed. Recall that r requires protein‑free RNA to bind to and to move along.
- b. A nonsense mutation before the cryptic r‑dependent terminator would cause the ribosomes to dissociate, now exposing the cryptic terminator in a protein‑free stretch of RNA. The hexamer r can bind and move along the RNA, and when it encounters an RNA polymerase stalled, or paused, at the terminator, it will cause the RNA polymerase to dissociate and the RNA to be released, hence preventing transcription of the subsequent genes in the operon.
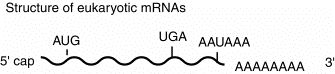
This general structure is true for almost all eukaryotic mRNAs. The cap structure is almost ubiquitous. A few examples of mRNAs without poly A at the 3' end have been found. Some of the most abundant mRNAs without poly A encode the histones. However, most mRNAs do have the 3' poly A tail.
The poly A tail at the 3' end can be used to purify mRNAs from other RNAs. Total RNA from a cell (which is about 90% rRNA and less than 10% mRNA) can be passed over an oligo(dT)-cellulose column. The poly A-containing mRNAs will bind, whereas other RNAs will elute.